In a study published in the International Journal of Molecular Sciences, researchers Dr. Ádám Sturm and Dr. Tibor Vellai from Eötvös Loránd University have made a significant leap forward in our understanding of the aging process. Their latest research reveals a hidden epigenetic mechanism in mitochondrial DNA (mtDNA) that could revolutionize how we approach aging research and diagnostics.
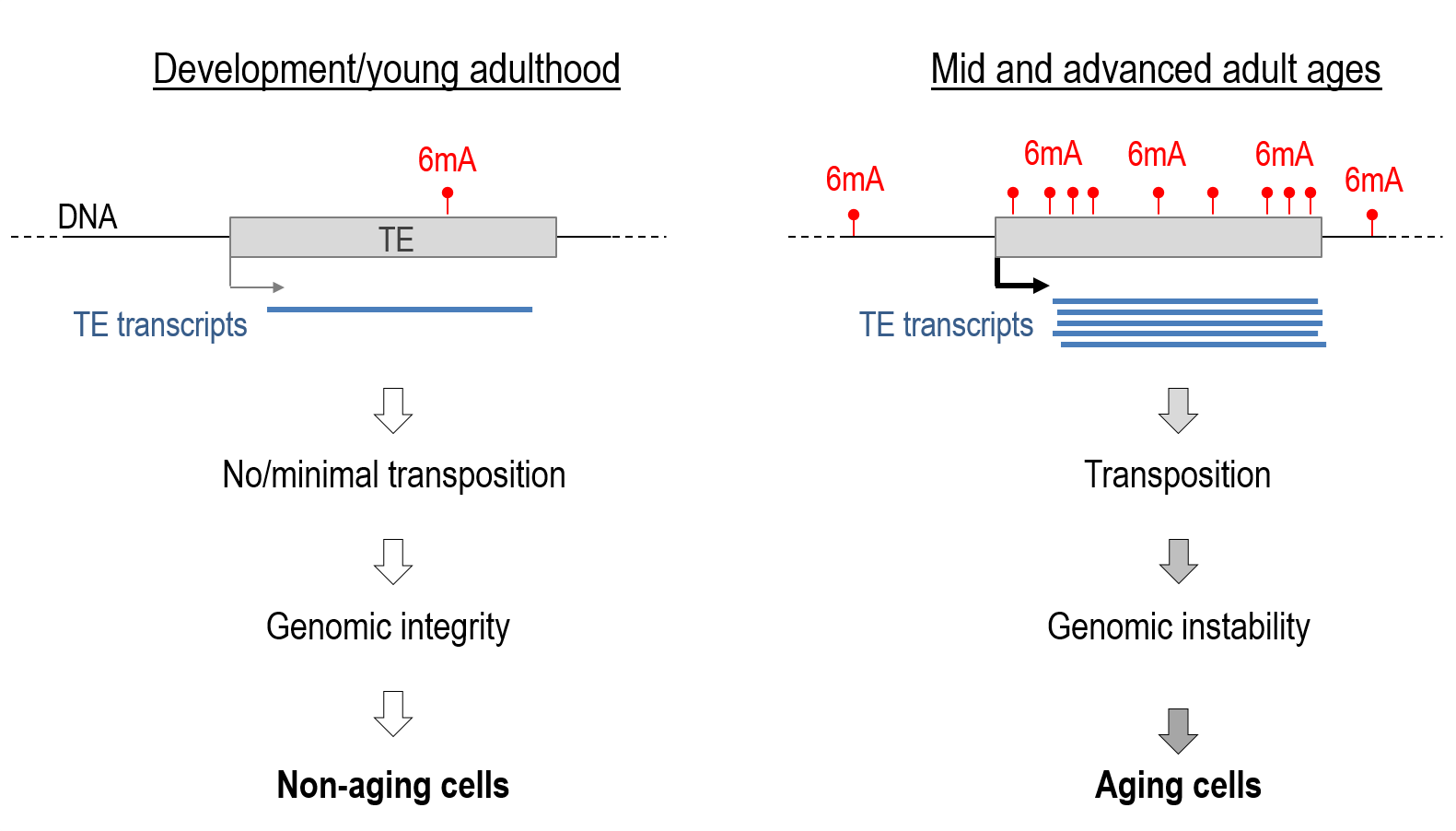
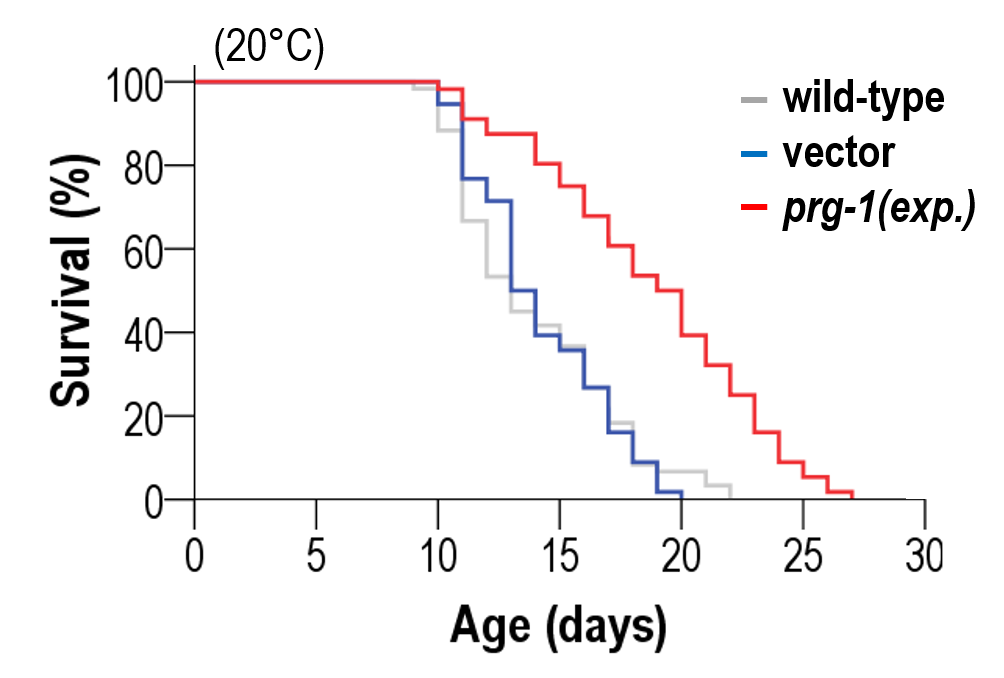
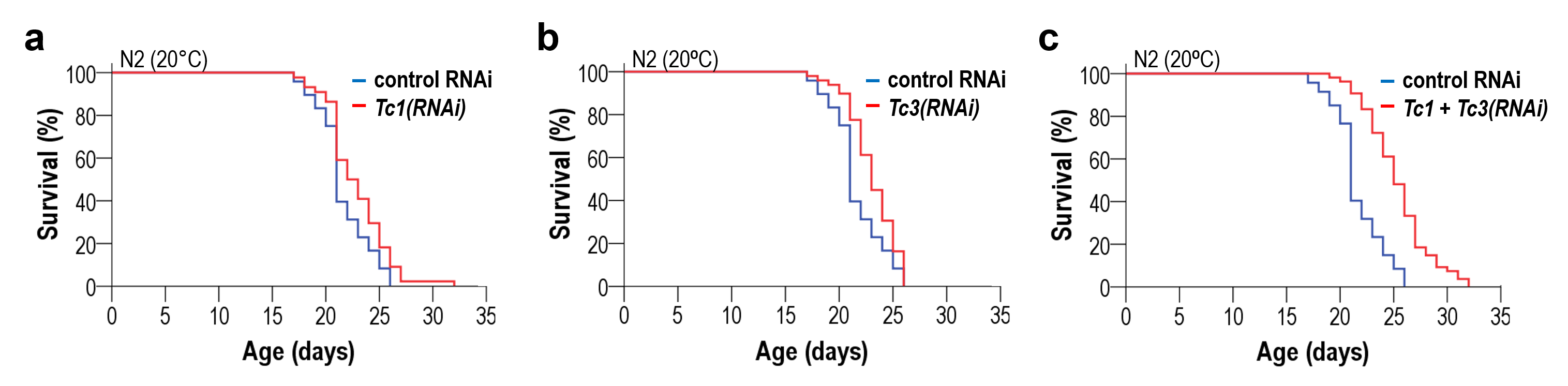
This new discovery builds upon the researchers’ previous landmark studies. In 2015, they published “The mechanism of aging: primary role of transposable elements in genome disintegration,” which established the crucial role of transposable elements in the aging process. They followed this up in 2023 with “Downregulation of transposable elements extends lifespan in Caenorhabditis elegans,” further solidifying the connection between transposable elements and longevity.
The Mitochondrial Epigenetic Clock
The current study unveils a previously hidden DNA modification called N6-methyladenine (6mA) that accumulates progressively in mtDNA as organisms age. This phenomenon was observed across diverse species, including the nematode Caenorhabditis elegans, the fruit fly Drosophila melanogaster, and dogs. The consistency across these different species suggests an evolutionarily conserved mechanism in the aging process that could potentially apply to all animal species, including humans.
Dr. Sturm describes this discovery as a “mitochondrial epigenetic clock.” This clock ticks at different rates depending on the lifespan of the organism, providing a new perspective on how aging is regulated at the cellular level. The implications of this discovery are far-reaching, offering new insights into the molecular mechanisms of aging and potential avenues for intervention.
Overcoming Methodological Challenges
One of the key achievements of this study was the development of a novel, reliable PCR-based method for detecting 6mA modifications. This technique allows for accurate, sequence-specific measurement of 6mA levels in mtDNA, overcoming limitations of previous methods that had led to disputes about the existence of 6mA in animal genomes.
The researchers’ method relies on PCR-directed amplification of target sequences digested by a 6mA-dependent restriction enzyme and ligated to a linker stretch. This approach eliminates artifacts due to detection noise or bacterial contamination, providing unambiguous identification of 6mA sites in mitochondrial (and potentially nuclear) genomes.

Figure1. The new PCR based 6ma quantification method. Detailed description here.
Linking 6mA Accumulation to Lifespan
A particularly intriguing finding of the study was that long-lived C. elegans mutants, which live twice as long as wild-type worms, accumulate 6mA at half the rate of their normal counterparts. This observation strongly links the rate of 6mA accumulation to the aging process and lifespan regulation. The researchers propose that N6-adenine methylation may serve as a mitochondrion-specific epigenetic clock. The gradual increase in mtDNA 6mA levels during aging is so robust and evolutionarily conserved that it can serve as a reliable epigenetic mark to determine age from a given tissue sample.
Single-Molecule Real-Time (SMRT) Sequencing Analysis
The researchers performed SMRT sequencing analysis on mtDNA samples isolated from C. elegans at different adult stages, specifically days 1 and 5. This technique allows for the direct detection of DNA modifications, including N6-methyladenine, during the sequencing process.
The results of the SMRT sequencing were striking. By identifying N6-methylated adenine sites and comparing the total 6mA levels of each chromosome with those of the mitochondrial genome within each age group, the team observed that N6-adenine methylation in the mtDNA relative to the nuclear genome was more robust in the aged sample.

Figure 2. SMRTseq analyses confirm that N6-methyadenine levels in the C. elegans mtDNA increase with age.
Implications for Aging Research and Diagnostics
The discovery of this mitochondrial epigenetic clock opens up new avenues for understanding and potentially intervening in the aging process. Dr. Vellai emphasizes that this epigenetic clock in mtDNA could serve as a more accessible and cost-effective way to measure biological age, compared to existing methods.
Current age-determining biomarkers and methods, such as Horvath’s clock (based on cytosine methylation), transcriptome-based aging clocks, inflammatory aging clocks, and metabolomic aging clocks, while informative, are often expensive and time-consuming. In contrast, the mtDNA 6mA-detecting PCR-based method presented in this study is relatively inexpensive and fast to perform, providing an opportunity for widespread use in scientific research, clinical diagnostics, and even forensics.
The Lifespan Limit Line
One of the most intriguing findings of the study is the discovery of what the researchers call a “Lifespan Limit Line.” This represents a point where the epigenetic marker (6mA) in mitochondrial DNA reaches a peak before declining, signaling the rapid onset of the aging process with severe health deteriorations.
This concept of a Lifespan Limit Line could have profound implications for our understanding of the maximum potential lifespan of different species and individuals. It may also provide insights into the biological processes that occur as organisms approach the end of their natural lifespan.

Figure 3. Discovery of a Lifespan Limit Line across species. The figure illustrates a surprising finding of a “Lifespan Limit Line”—a point where an epigenetic marker in mitochondrial DNA reaches a peak before declining, signaling the rapid onset of the aging process with severe health deteriorations.
Future Directions
The study paves the way for future research on how environmental factors, lifestyle choices, and potential interventions might influence the rate of 6mA accumulation in mtDNA. Understanding these epigenetic changes could lead to novel strategies for promoting healthier aging and potentially extending healthspan.
Moreover, the connection between this newly discovered epigenetic clock and the researchers’ previous work on transposable elements opens up intriguing questions about the interplay between different cellular and molecular mechanisms in the aging process.